Research

Simulating the dynamics of large molecules has been one of the key goals of theoretical chemistry. Although classical molecular dynamics has been successful in many cases, it falls short in accounting for quantum-mechanical effects, such as tunnelling and nonadiabatic transitions. It is known that these effects can not only dramatically affect the rate of a chemical reaction but also change the reaction mechanism.
In principle, fully quantum-mechanical methods give exact results, but in practice, they are only applicable to small molecules or simplified models. The key challenge is thus to account for quantum-mechanical behaviour for large complex systems with a computational efficiency similar to that of classical approaches.
Semiclassical instanton theory
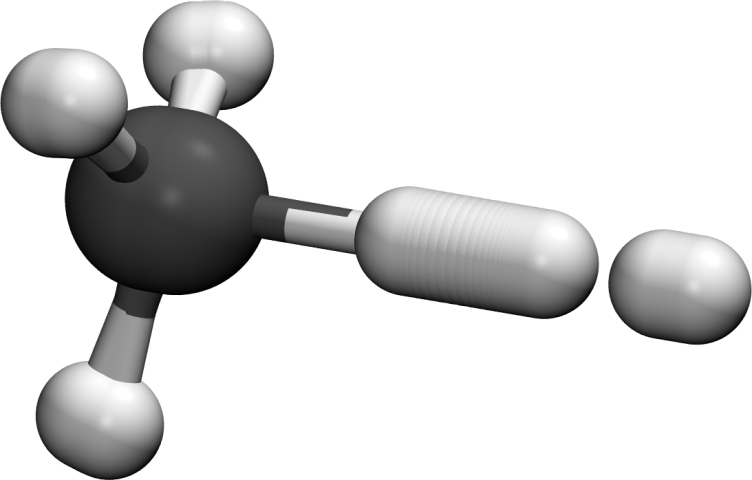
Calculating low temperature rates of polyatomic chemical reactions
Tunnelling in water clusters and other hydrogen-bonded molecular systems
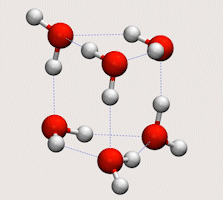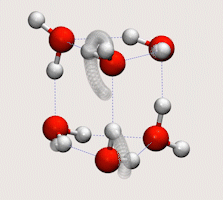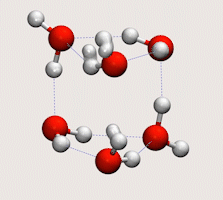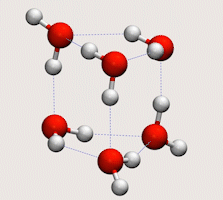
Simulating tunnelling dynamics in molecular clusters
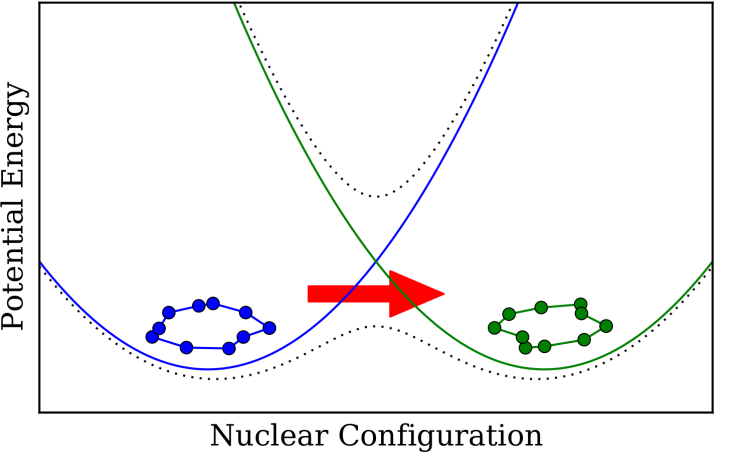
Nonadiabatic Rate Theory

Extending instanton theory to nonadiabatic processes such as Fermi's golden rule and beyond
